Published Date: July 7, 2025
Topics Included: Innovative Trials, Regulatory Submissions + Approvals
Share to:
Introduction
The following recommendations aim to highlight the unique benefits of using disease progression modeling in medical product development, when it should be considered over other tools or approaches, and provide considerations for what is needed and how to incorporate it.
These recommendations are intended for decision makers in medical product development, including chief medical officers, heads of innovation or translational medicine, leadership roles in therapeutic areas, project team leads, and regulatory affairs.
Cross functional leaders often encounter similar questions around internal resources, evidence prioritization, and return of investment when making medical product development decisions. These recommendations will help decision makers ask the right questions of modeling experts and other subject matter experts to recognize the value of DPM and inform decisions about its implementation.
What is disease progression modeling?
A disease progression model (DPM) is a mathematical model that quantitatively describes the time course or trajectory of a disease.
Disease progression modeling can link disease progress, treatment effect, and/or patient behavior to clinical trial outcomes and inform decision making throughout the medical product development process.
While some modeling and simulation approaches such as pharmacokinetic/pharmacodynamic (PK/PD) and physiologically based pharmacokinetic (PBPK) modeling are well recognized as enabling clinical trials and model-informed drug development, the potential of disease progression modeling to improve the quality and efficiency of medical product development has yet to be fully realized.
How can disease progression modeling impact medical product development?
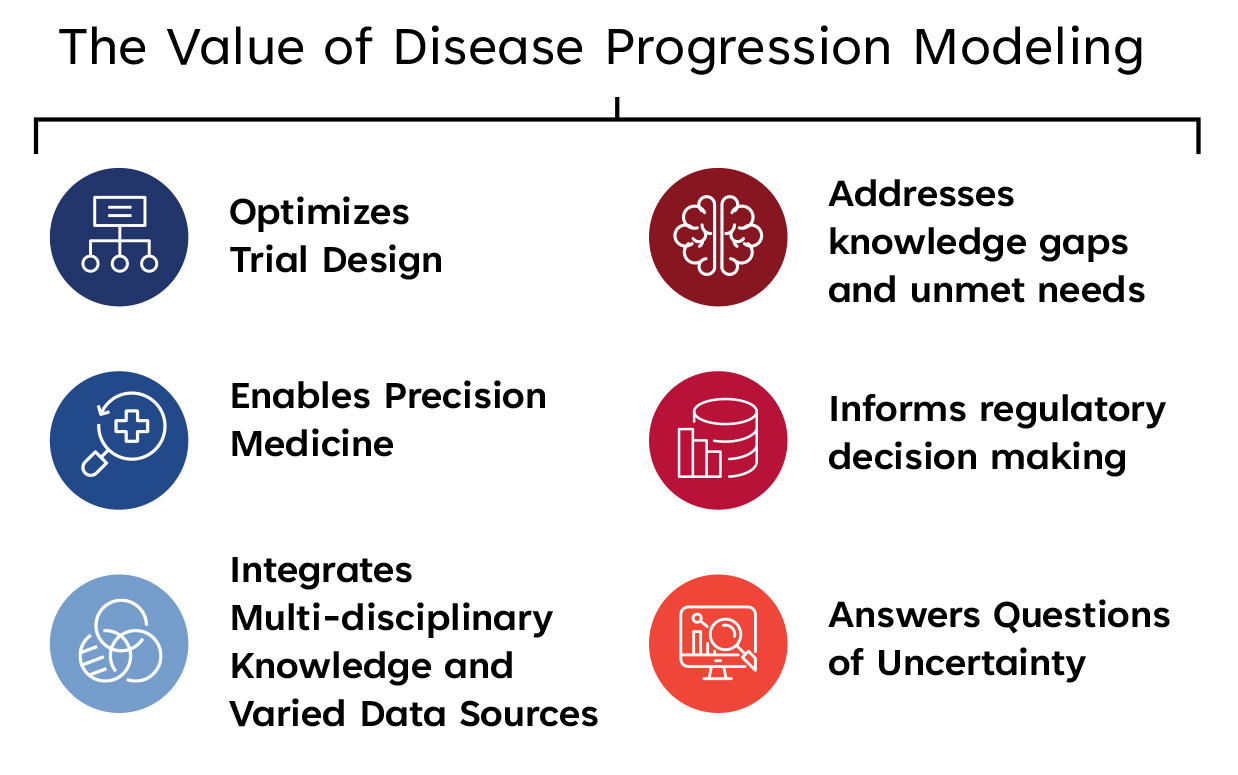
The use of a DPM integrates multi-disciplinary knowledge and data from different sources, including translational, clinical trial, and real world data to:
- Impact trial efficiency, especially for trials with diseases that progress over a long duration of time, for which running a clinical trial during that period of time is costly and burdensome.
- Answer questions of uncertainty. It can account for and provide clarity around heterogeneity across patients and at an individual level.
- Tailor trials towards precision medicine. Recognizing that diseases progress differently, across populations and in different stages within a patient, DPM can address individual factors or covariates affecting progression to enable trial design optimization and tailor trials towards precision medicine (e.g. stratify populations or adapt treatment plans).
- Address unmet needs and knowledge gaps, including supplementing for a small population size with digital twins in rare disease trials, or stratifying a population for patient enrichment for diseases with heterogeneous phenotypes (e.g. autoimmune and neurodegenerative diseases), or supporting a limited understanding of the disease by modeling disease course for endpoint selection.
- Inform regulatory decision making. DPM contributes to the totality of understanding of a medical product’s benefits and risks, helping to advance its development and support regulatory decision-making both in premarket product review and post-market product assessment.
CTTI further describes the value of DPM, its current applications, and opportunities for uptake in the following article published in Clinical Pharmacology & Therapeutics called The Potential of Disease Progression Modeling to Advance Clinical Development and Decision Making.
Recommendations Summary
- Evaluate if longitudinal data about the disease course is critical to answer a question of interest for medical product development decisions.
- Assess the value of using a disease progression model compared with other potential approaches.
- Use the most up-to-date understanding of the disease and measure(s) of progression available to inform your disease progression modeling approach.
- Determine whether the necessary sources of data are available, relevant and reliable to develop a disease progression model.
- Determine whether technological and model resources, as well as the right skill sets, exist or can be obtained to support the disease progression model development and implementation.
- Begin using disease progression modeling early on in the medical product development lifecycle to build confidence in the model for later stages of development.
- Continually assess the performance and ensure the disease progression model is qualified for the intended application.
- Leverage new technology and evolving methods to advance uses of disease progression models.
- Disseminate critical insights on disease progression modeling to promote standardized practices and foster confidence.
Recommendations
Future Directions
Ongoing advancements in technology, such as the ability to leverage continuous data from digital health technologies and the potential for synthetic data, coupled with regulatory support and resources (e.g., via FDA’s Quantitative Medicine and Digital Health Centers of Excellence), present significant opportunities for future developments in disease progression modeling (DPM).
While the majority of DPM work has traditionally focused on drug development, these models can also support device development and drug-device combinations. Notable examples include the use of modeling for artificial pancreas systems and brain stimulation treatments for Parkinson’s disease. Therefore, the recommendations within this document are designed to be applicable across various contexts of medical product development.
Moreover, disease progression models can aid in forecasting clinical response trajectories in healthcare settings beyond drug development, offering a framework for integration with clinical practice. This integration can enhance patient care by providing more accurate predictions of disease progression and treatment outcomes.
CTTI encourages consortia and public-private partnerships to actively track these opportunities and monitor how they may improve the quality and efficiency of clinical trials and medical product development. By fostering collaboration and innovation, we can ensure that disease progression modeling continues to evolve and contribute to advancements in healthcare.
Methods
Experts from across the clinical trials ecosystem developed these recommendations following CTTI’s five-step methodology design to ensure the recommendations are actionable, evidence-based, and consensus driven.
Acknowledgements:
Thank you to the experts and key contributors from across the clinical trials ecosystem who helped create this set of recommendations and resources, including the Disease Progression Modeling project team leaders and members, Expert Meeting participants, Recommendations Advisory Committee members, and many others.
Table 1.
Appendix
Examples of DPM use for medical product development decision making:
Regulatory:
- Goteti, K. Opportunities and Challenges of Disease Progression Modeling in Drug Development – An IQ Perspective. Clin Pharmacol Ther. Feb 18 2023; https://doi.org/10.1002/cpt.2873
- Disease models listed in Table 2 on FDA’s Division of Pharmacometrics webpage
- EMA’s qualification responses (based on specific disease areas):
- Qualification opinion of a novel data driven model of disease progression and trial evaluation in mild and moderate Alzheimer’s disease
- Qualification Opinion of Islet Autoantibodies (AAs) as Enrichment Biomarkers for Type 1 Diabetes (T1D) Prevention Clinical Trials qualification-opinion- prognostic- covariate-adjustment-procovatm_en.pdf
- Letter of Support of Model-based Clinical Trial Simulation Platform (CTSP) for Duchenne Muscular Dystrophy letter-support model-based- clinicaltrial-simulation-platform-optimizedesignefficacy-evaluation-studies- parkinsonsdisease_en.pdf
- Letter of support for “Islet autoantibodies as enrichment biomarkers for type 1 diabetes prevention studies, through a quantitative disease progression model”
- Letter of support for Model-based CT enrichment tool for CTs in aMCI
DPM used to stratify a population to inform treatment or outcome measures and optimize trial design:
- Romero, K. et al. Molecular neuroimaging of the dopamine transporter as a patient enrichment biomarker for clinical trials for early Parkinson’s disease. Clin. Transl. Sci. 12, 240-246 (2019)
- Feng Y, Wang X, Suryawanshi S, Bello A, Roy A. Linking tumor growth dynamics to survival in ipilimumab-treated patients with advanced melanoma using mixture tumor growth dynamic modeling. CPT Pharmacometrics Syst Pharmacol. Nov 2019; 8(11):825–834. doi:10.1002/psp4.12454
DPM used for a rare disease with data paucity:
- Reetz K, Dogan I, Hilgers RD, et al. Progression characteristics of the European Friedreich’s ataxia consortium for translational studies (EFACTS): A 4-year cohort study. Lancet Neurol. May 2021;20(5):362-372. doi:10.1016/ S1474-4422(21)00027-2
DPM used to link pathophysiology to medical product development decisions:
- Kaddi, C.D. et al. Quantitative systems pharmacology modeling of acid sphingomyelinase deficiency and the enzyme replacement therapy olipudase alfa is an innovative tool for linking pathophysiology and pharmacology. CPT Pharmacometrics Syst. Pharmacol. 7, 442-452 (2018).
Resources for the use of data for DPM development and implementation:
- Regulatory resources:
- FDA Real World Evidence (RWE) webpage that includes relevant guidance documents on the use of real-world data
- Blacketer C, Schuemie FJ, Ryan PB, Rijnbeek P (2021). “Increasing trust in real-world evidence through evaluation of observational data quality.” Journal of the American Medical Informatics Association, 28(10), 2251-2257. https://doi.org/10.1093/jamia/ocab132
- General trial data resources:
Examples of DPM used to bridge gaps between early and late stage development phases to inform early go/no go decisions:
- Wang, Y. et al. Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin. Pharmacol. Ther. 86, 167-174 (2009).
- Sheng Y, Teng SW, Wang J, Wang H, Tse AN. Tumor growth inhibition-overall survival modeling in non-small cell lung cancer: A case study from GEMSTONE-302. CPT Pharmacometrics Syst Pharmacol. 2024 Mar;13(3):437-448. doi: 10.1002/psp4.13094. Epub 2023 Dec 21. PMID: 38111189; PMCID: PMC10941555.
Example resources to assess the performance of a DPM:
- General Principles for Model-Informed Drug Development. ICH Harmonised Guideline
- Toward Good Simulation Practice
- EFPIA MID3 Workgroup (2016). Good practices in model-informed drug discovery and development (MID3): Practice, application and documentation. CPT: Pharmacometrics & Systems Pharmacology (5) 93-122.doi: 10.1002/psp4.12049
- Musuamba et al. 2021 Scientific and regulatory evaluation of mechanistic in silico drug and disease models in drug development: Building model credibility CPT-PSP (10), 804
- Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions Guidance for Industry and Food and Drug Administration Staff.
- ASME’s V&V 40 Assessing Credibility of Computational Modeling through Verification and Validation: Application to Medical Devices
Machine Learning in DPM used to enhance precision medicine:
- Terranova N, Venkatakrishnan K. Machine learning in modeling disease trajectory and treatment outcomes: An emerging enabler for model-informed precision medicine. Clin Pharmacol Ther. Dec 17 2023; doi:10.1002/cpt.3153
Collaborative Groups and Case Study Exchanges:
- The Clinical Trials Transformation Initiative’s Case Studies Exchange
- Case studies from
- FDA's
- Critical Path Tools and Platforms
References
- S, Nicholas T, Azer K, Corrigan BW. Role of disease progression models in drug development. Pharm Res. Aug 2022;39(8): 1803-1815. doi:10.1007/s11095-022-03257-3
- Starling, S. The Potential of Disease Progression Modeling to Advance Clinical Development and Decision Making Clin Pharmacol Ther. Oct 15 2024; https://doi.org/10.1002/cpt.3
- Madabushi R, Benjamin J, Zhu H, Zineh I. The US Food and Drug Administration’s model-informed drug development meeting program: From pilot to pathway. Clin Pharmacol Ther. Mar 6 2024; doi:10.1002/cpt.3228
- Barrett, J.S., Betourne, A., Walls, R.L. et al. The future of rare disease drug development: the rare disease cures accelerator data analytics platform (RDCA-DAP). J Pharmacokinet Pharmacodyn 50, 507–519 (2023). https://doi.org/10.1007/s10928-023-09859-7
- Sheng Y, Teng SW, Wang J, Wang H, Tse AN. Tumor growth inhibition-overall survival modeling in non-small cell lung cancer: A case study from GEMSTONE-302. CPT Pharmacometrics Syst Pharmacol. 2024 Mar;13(3): 437-448. doi: 10.1002 psp4.13094. Epub 2023 Dec 21. PMID: 38111189; PMCID: PMC109
- Corneli A, Hallinan Z, Hamre G, et al. The clinical trials transformation initiative: Methodology supporting the mission. Clin Trials. Feb 2018;15 (1_suppl):13-18. doi:10.1177/1740774518755054.” psp4.13094. Epub 2023 Dec 21. PMID: 38111189; PMCID: PMC10941555.
- Aghamiri SS, Amin R, Helikar T. Recent applications of quantitative systems pharmacology and machine learning models across diseases. J Pharmacokinet Pharmacodyn. 2022 Feb;49(1):19-37. doi: 10.1007/s10928-021-09790-9. Epub 2021 Oct 20. PMID: 34671863; PMCID: PMC8528185.
- Myles, Puja, Mitchell, Colin, Redrup Hill, Elizabeth, Foschini, Luca and Wang, Zhenchen (2024, June 1). High-fidelity synthetic patient data applications and privacy considerations. In the Journal of Data Protection & Privacy, Volume 6, Issue 4. https://doi.org/10.69554/LQOM5698.