 Patient and Caregiver
Patient and Caregiver Investigator
Investigator Regulator
Regulator IRB
IRB Tech, Device,
Tech, Device, Payer
PayerCollaborative Engagement in Clinical Trial Design
How to Use This Roadmap
This roadmap is designed to support clinical trial designers, sponsors, and program teams as they move from study concept through final protocol development. t provides stage-specific guidance on how to engage key collaborators—including patients and caregivers, investigators and site staff, regulatory agencies, institutional review boards or ethics committees (IRB/IEC), technology and data partners, and payers—to strengthen trial design and execution.
For each stage of trial design, it explains:
- Why engaging specific collaborator groups matters at that point
- What types of insights those collaborators can provide
- How sponsors can engage them effectively using practical strategies
This guide focuses on engagement during protocol development stages (study concept, synopsis, draft protocol, and final protocol), with the exception of regulatory engagement, which occurs iteratively across early, mid, and late stages of development.
By aligning collaborative engagement with Quality by Design principles, the roadmap helps teams design trials that are not only scientifically sound, but also feasible to conduct, meaningful to participants, responsive to regulatory and payer expectations, and positioned to deliver real‑world impact.
A Quality‑by‑Design Approach to Collaborative Trial Design
Designing high‑quality clinical trials is increasingly complex. Sponsors must deliver studies that are scientifically rigorous, operationally executable, ethically sound, and capable of generating evidence that supports regulatory approval, real‑world use, and patient access. Achieving all of this requires more than technical excellence—it requires intentional collaboration across the clinical trial ecosystem.
The Collaborative Engagement in Clinical Trial Design Roadmap was developed to help navigate this complexity by providing practical, phase‑specific guidance on engaging key collaborators throughout the trial design process. These collaborators—including patients and caregivers, investigators and site staff, regulatory agencies, institutional review boards / ethics committees (IRB/IEC), technology and data partners, and payers—bring distinct perspectives that strengthen study design, surface risks earlier, and improve the likelihood of successful trial execution.
At its core, this roadmap is grounded in Quality by Design (QbD). QbD emphasizes identifying what is truly critical to quality and proactively designing trials to reliably deliver meaningful outcomes. While QbD principles are well established, sponsors often lack clear guidance on when to engage different collaborator groups, why their input matters at specific stages, and how to incorporate that input in a practical and efficient way. This roadmap addresses that need.
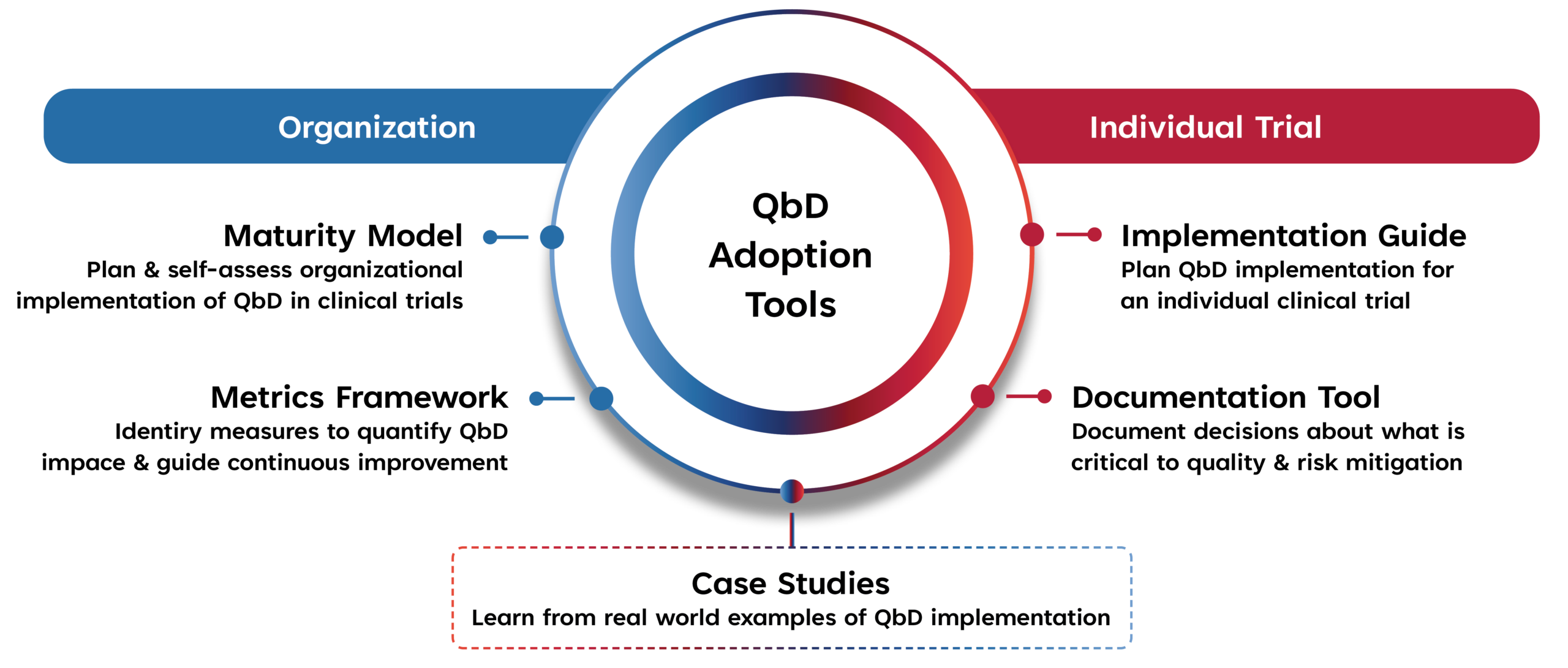
Bringing Quality by Design to Life
Quality by Design is most effective when trial designs are intentionally “pressure‑tested” through multiple perspectives. Different collaborators surface different types of risks—and that diversity of insight is a strength of the process.
- Sponsors and CROs often identify systemic and design‑level risks, such as vulnerabilities that could affect study validity, endpoint interpretability, or data integrity.
- Clinical sites provide essential insight into operational feasibility—how protocols function within real clinic workflows, patient populations, and resource constraints.
- Patients and clinical experts highlight feasibility, burden, and ethical considerations that influence enrollment, retention, and overall trial experience.
By encouraging open dialogue and critical thinking across these perspectives, trial teams can identify “paper versus practice” gaps early, simplify overly complex designs, and focus resources on what truly matters.
A QbD‑driven, collaborative approach also promotes leaner, more purposeful trials. Every procedure, assessment, and data point should serve a clear objective related to participant safety or meaningful endpoints. Eliminating non‑essential complexity reduces burden, minimizes opportunities for error, and supports data credibility.
Collaborator Engagement
It is important to note that while the focus of this specific roadmap is on the process of designing a clinical trial -- from concept to final protocol -- collaborator engagement in drug development must start much earlier than the protocol-writing stage. To make a truly meaningful impact sponsors must engage with diverse collaborators as early as possible to ensure they have a strong understanding of the current treatment landscape, physician behaviors and preferences, outcomes and benefits patients desire in their care, as well as the gaps their new medicine will address. Early and systematic engagement is a cornerstone of purposeful drug development. As a result, the Collaborative Engagement in Clinical Trial Design includes background documents that outline various stages at which collaborator input should be sought prior to protocol writing. Sponsors should consult these documents to develop a robust engagement strategy for their drug development programs and associated clinical trials. While each study may be unique in some ways and is fit for purpose, all studies should share in the early and often engagement of diverse collaborator groups and be written and edited with a quality approach.